Article Text

Abstract
Objective To determine whether once-daily esomeprazole 40 mg or 20 mg compared with placebo reduces the incidence of peptic ulcers over 26 weeks of treatment in patients taking low-dose acetylsalicylic acid (ASA) and who are at risk for ulcer development.
Design Multinational, randomised, blinded, parallel-group, placebo-controlled trial.
Setting Cardiology, primary care and gastroenterology centres (n=240).
Patients Helicobacter pylori-negative patients taking daily low-dose ASA (75–325 mg), who fulfilled one or more of the following criteria: age ≥18 years with history of uncomplicated peptic ulcer; age ≥60 years with either stable coronary artery disease, upper gastrointestinal symptoms and five or more gastric/duodenal erosions, or low-dose ASA treatment initiated within 1 month of randomisation; or age ≥65 years. All patients were ulcer-free at study entry.
Interventions Once-daily, blinded treatment with esomeprazole 40 mg, 20 mg or placebo for 26 weeks.
Main outcome measures The primary end point was the occurrence of endoscopy-confirmed peptic ulcer over 26 weeks.
Results A total of 2426 patients (52% men; mean age 68 years) were randomised. After 26 weeks, esomeprazole 40 mg and 20 mg significantly reduced the cumulative proportion of patients developing peptic ulcers; 1.5% of esomeprazole 40 mg and 1.1% of esomeprazole 20 mg recipients, compared with 7.4% of placebo recipients, developed peptic ulcers (both p<0.0001 vs placebo). Esomeprazole was generally well tolerated.
Conclusions Acid-suppressive treatment with once-daily esomeprazole 40 mg or 20 mg reduces the occurrence of peptic ulcers in patients at risk for ulcer development who are taking low-dose ASA.
Clinical trial registration number ClinicalTrials.gov identifier: NCT00441727.
- Peptic ulcer
- esomeprazole
- acetylsalicylic acid
- cardiovascular risk management
- clinical trials
This is an open-access article distributed under the terms of the Creative Commons Attribution Non-commercial License, which permits use, distribution, and reproduction in any medium, provided the original work is properly cited, the use is non commercial and is otherwise in compliance with the license. See: http://creativecommons.org/licenses/by-nc/2.0/ and http://creativecommons.org/licenses/by-nc/2.0/legalcode.
Statistics from Altmetric.com
Introduction
For patients at increased cardiovascular risk, continuous low-dose acetylsalicylic acid (ASA, aspirin; 75–325 mg/day) reduces the risk of a range of cardiovascular events.1 2 Low-dose ASA is a mainstay of protective treatment for secondary prevention of cardiovascular events, and is recommended for primary prevention in patients at high cardiovascular risk.3 4 In the most recent meta-analysis conducted by the Antithrombotic Trialists' Collaboration, low-dose ASA was associated with a significantly lower risk of serious vascular events (eg, myocardial infarction, stroke or vascular death) in secondary cardiovascular prevention trials.1 In primary prevention trials, a significant reduction in serious vascular events was due mainly to a reduction in non-fatal myocardial infarction, whereas stroke and vascular mortality were not significantly reduced. The benefit in primary prevention trials was offset by a significant increase in the risk of major gastrointestinal and extracranial bleeding.
Even at low doses, ASA is associated with gastrointestinal adverse effects, particularly in patients who are at increased gastrointestinal risk such as older patients and those with a previous history of ulcer disease.5 Low-dose ASA-associated adverse gastrointestinal effects range from troublesome upper gastrointestinal problems such as dyspeptic symptoms and heartburn6 7 to serious peptic ulcer complications, including gastrointestinal bleeding6 8 and perforated ulcers.9 Peptic ulcer complications have been reported to occur more than twice as frequently among low-dose ASA users than controls,10 11 even among patients taking low-dose ASA for >3 months.12 The risk of gastrointestinal bleeding remains elevated with longer duration of ASA treatment13; indeed, the risk of gastrointestinal bleeding after 1 year of ASA use is more than double relative to non-users.11 Furthermore, the incidence of upper gastrointestinal bleeding has increased in parallel with the widespread use of low-dose ASA.14 Use of enteric-coated or buffered formulations of low-dose ASA may not mitigate the risk of gastrointestinal complications.10 Gastrointestinal bleeding can occur without preceding symptoms,15 and may lead to discontinuation of low-dose ASA treatment.16 Adverse upper gastrointestinal symptoms with low-dose ASA use may lead to poor adherence to, or discontinuation of, low-dose ASA treatment17; this is worrying as discontinuation is associated with a markedly increased risk of serious adverse cardiovascular events that can occur within a few weeks.18 Therefore, continuing to treat patients at cardiovascular risk with long-term low-dose ASA is key in the management of cardiovascular disease.19 For these reasons, concomitant gastroprotection with a proton pump inhibitor (PPI) is recommended for cardiovascular patients who require continuous low-dose ASA and are at an increased gastrointestinal risk.20
The OBERON trial explored primarily the efficacy of esomeprazole, at once-daily doses of 40 mg and 20 mg, compared with placebo, in the prevention of peptic ulcers associated with continuous use of low-dose ASA in patients who were at an increased risk of developing peptic ulcers.
Patients and methods
OBERON was a randomised, blinded, parallel-group, placebo-controlled trial (ClinicalTrials.gov identifier: NCT00441727; AstraZeneca study code: D961FC00003), conducted at 240 cardiology, primary care and gastroenterology centres in 20 countries (Argentina, Australia, Bulgaria, Canada, Czech Republic, Finland, Germany, Indonesia, Mexico, Norway, Philippines, Poland, Portugal, Romania, Russia, Slovakia, South Africa, South Korea, Thailand and USA) in accordance with the Declaration of Helsinki and Good Clinical Practice guidelines. Patients, healthcare providers, data collectors and outcome adjudicators were blinded to treatment allocation. The study protocol was approved by the relevant local institutional review boards or independent ethics committees of each study centre, according to local regulations. All patients provided written, informed consent before enrolment in the trial. The first patient was randomised to the study on 22 February 2007 and the last patient completed the study on 28 August 2008.
Patients
Patients whose doctor prescribed or recommended daily use (≥5 days/week) of low-dose ASA (75–325 mg/day) and who were Helicobacter pylori negative at screening were eligible to participate if they fulfilled one or more of the following inclusion criteria: aged ≥18 years with documented history of uncomplicated peptic ulcer; aged ≥60 years with one or more risk factor (stable coronary artery disease, or complaints of upper gastrointestinal symptoms that, as judged by the investigator, required an endoscopy resulting in a finding of five or more gastric and/or duodenal erosions at baseline endoscopy, or low-dose ASA-naïve (ie, treatment was initiated within 1 month of randomisation)); or aged ≥65 years. Patients at very high cardiovascular and/or gastrointestinal risk were excluded from the study for ethical reasons. Cardiovascular exclusion criteria were unstable hypertension; recent (within 3 months) experience of acute coronary syndromes, percutaneous coronary intervention, coronary artery bypass grafting, clinically relevant valvular disease, serious cardiac failure (New York Heart Association class II–IV or ejection fraction <40%) and stroke. Gastrointestinal exclusion criteria were Los Angeles grade C–D erosive (reflux) oesophagitis at baseline; patient-reported severe oesophagitis within 1 year; peptic ulcer at baseline; history of peptic ulcer complications (eg, clinically significant bleeding and/or perforation) and previous gastric or duodenal surgery (patients who had undergone laparoscopic fundoplication were eligible). Other exclusion criteria were unstable diabetes mellitus: continuous treatment with a non-steroidal anti-inflammatory drug within 2 months of randomisation; ongoing anticoagulant treatment (antiplatelets such as clopidogrel were permitted); use of drugs that interact with esomeprazole (phenytoin, ketoconazole, itraconazole, voriconazole, cisapride, atazanavir, ritonavir); use of a PPI or prostaglandin analogue within 14 days of baseline endoscopy, or between baseline endoscopy and randomisation; daily use of histamine 2-receptor antagonists within 14 days of baseline endoscopy; and need for continuous treatment with prostaglandin analogues or sucralfate.
H pylori tests were performed according to local routine practice and patients who were H pylori positive had to have completed eradication treatment ≥4 weeks before randomisation. Baseline H pylori status was subsequently confirmed by a [13C]urea breath test and analysed by a central laboratory (Quintiles, Research Triangle Park, North Carolina, USA); confirmation test results were blinded to the investigator during the course of the study.
Treatment and design
After endoscopy and in addition to the low-dose ASA regimen prescribed by their doctor, eligible patients were randomised sequentially by numbered, opaque coded envelopes (in blocks of six) to 26 weeks' oral treatment with esomeprazole capsules at once-daily doses of 40 mg or 20 mg, or placebo (in a ratio of 1:1:1). The randomisation codes were assigned from a computer-generated list held by the study sponsor (AstraZeneca R&D, Mölndal, Sweden); code envelopes were used as an emergency unblinding tool. Esomeprazole and placebo capsules were supplied by the study sponsor and were identical in appearance and packaged in identical containers. Patients were instructed to take the study drug in the morning before breakfast with a glass of water. A rescue drug (antacids with acid-binding capacity of <16 mmol HCl/tablet) was provided for use as needed (≤6 tablets/day). Patients returned used study drug containers, rescue drug containers and all unused drugs to the study personnel at each study visit for assessment of drug adherence and rescue-drug use.
Outcomes
The primary end point was endoscopy-confirmed peptic (gastric or duodenal) ulcer during the 26-week treatment period. An ulcer was defined as a mucosal break measuring ≥3 mm over its largest diameter (size confirmed with endoscopy forceps) with a base (smooth or regular punched-out defect in the mucosa) and margin (discrete, sharply demarcated and usually raised in relation to the base) and the absence of any malignancy features. Endoscopy for investigation of peptic ulcers was performed at baseline, and at weeks 8 and 26 or upon withdrawal.
Secondary end points included the occurrence of a gastric ulcer and, separately, a duodenal ulcer, over 26 weeks of treatment, and safety and tolerability of treatments. Safety and tolerability during 26 weeks of treatment with esomeprazole or placebo were evaluated by assessments of adverse events and vital signs (including blood pressure and pulse rate), and by monitoring standard clinical laboratory tests and physical examinations. Spontaneously reported adverse events and those reported in response to a standardised question were recorded at all scheduled (weeks 8, 16 and 26) and unscheduled visits.
Statistical analyses
The primary analysis for the efficacy variables was based on the intention-to-treat (ITT) population, defined as all patients who were randomised. A supportive analysis for the primary end point was also conducted for the per-protocol population, which consisted of those patients in the ITT population with no major protocol violations, such as deviation from the inclusion/exclusion criteria, or insufficient intake of the study drug.
The cumulative incidence of peptic, duodenal and gastric ulcer(s) was estimated from Kaplan–Meier life tables, and treatment groups were compared with placebo using the log-rank test stratified by ASA dose (75–100 mg and 101–325 mg). Patients who did not complete the 26 weeks for other reasons than the development of an ulcer were censored at the time of study discontinuation. Peptic, gastric and duodenal ulcers were tested in a hierarchical closed-test procedure, which was performed in parallel for the two esomeprazole doses with adjustment for multiplicity, using the Hochberg procedure, in each step. The effect of esomeprazole treatment on peptic ulcer incidence, stratified by ASA dose (75–100 mg and 101–325 mg), was analysed post hoc using Fisher's exact test. Analyses were conducted using SAS software (version 8.2; SAS Institute Inc).
Sample-size calculation
Based on data from a previous trial21 and the assumption that this study population had a higher gastrointestinal risk, we assumed an event rate for peptic ulcers in the placebo group and esomeprazole recipients of 8.0% and 3.2%, respectively, corresponding to a relative risk reduction (RRR) of 60%. We also assumed that 70% of the ulcers would be gastric. A sample size of 2400 patients (800 per treatment arm) was selected to provide 90% power for the primary end point (two-sided α=0.05) at the 2.5% significance level, assuming a drop-out rate of 15%.
Results
Patients
A total of 2426 patients were randomised; all randomised patients were included in the ITT population as follows: esomeprazole 40 mg (n=817), esomeprazole 20 mg (n=804) or placebo (n=805) (figure 1). Among the randomised patients, few (3.4%) received concomitant clopidogrel treatment at any time during the study. Patient baseline characteristics were similar between treatment groups (table 1).
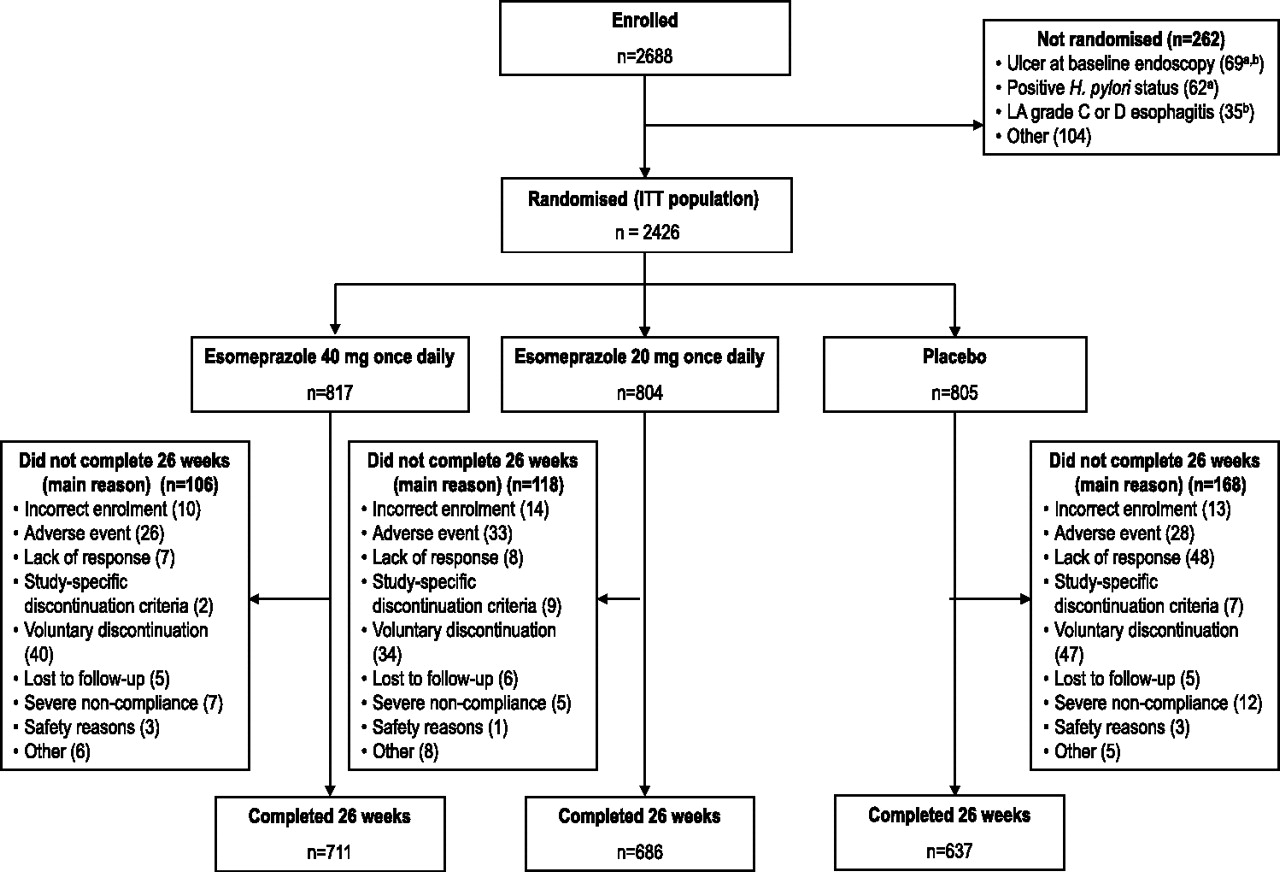
Patient flow through the study. All randomised patients are included in the intention-to-treat (ITT) analyses, including those patients who did not complete 26 weeks of treatment. Lack of response was defined as development of gastric and/or duodenal ulcer and/or upper gastrointestinal symptoms requiring active intervention; the study-specific discontinuation criterion was defined as low-dose acetylsalicylic acid (ASA) treatment permanently stopped; severe non-compliance was defined as all other forms of non-compliance except stopping low-dose ASA treatment; safety reasons were defined as those to protect study subjects from potential safety risks—for example, new data causing study termination or subpopulation of subjects to be discontinued. All discontinuations were judged by the study investigators. aTwo patients had positive Helicobacter pylori status and ulcer at baseline endoscopy; bsix patients had Los Angeles(LA) grade C or D erosive (reflux) oesophagitis and ulcer at baseline endoscopy.
Patient baseline characteristics (intention-to-treat population)
The per-protocol population included 1534 patients (esomeprazole 40 mg, 525 patients; esomeprazole 20 mg, 526 patients; placebo, 483 patients). The main reason for exclusion from the per-protocol population in all treatment groups was a positive H pylori test at study conclusion or missing H pylori status. The safety analysis included all patients who received one or more dose of investigational treatment and provided any follow-up data.
The majority (94%) of patients were adherent (took ≥75% of doses) to esomeprazole. Patients used their own prescribed or recommended low-dose ASA tablets on ≥5 days/week, which were not counted.
Incidence of ulcers
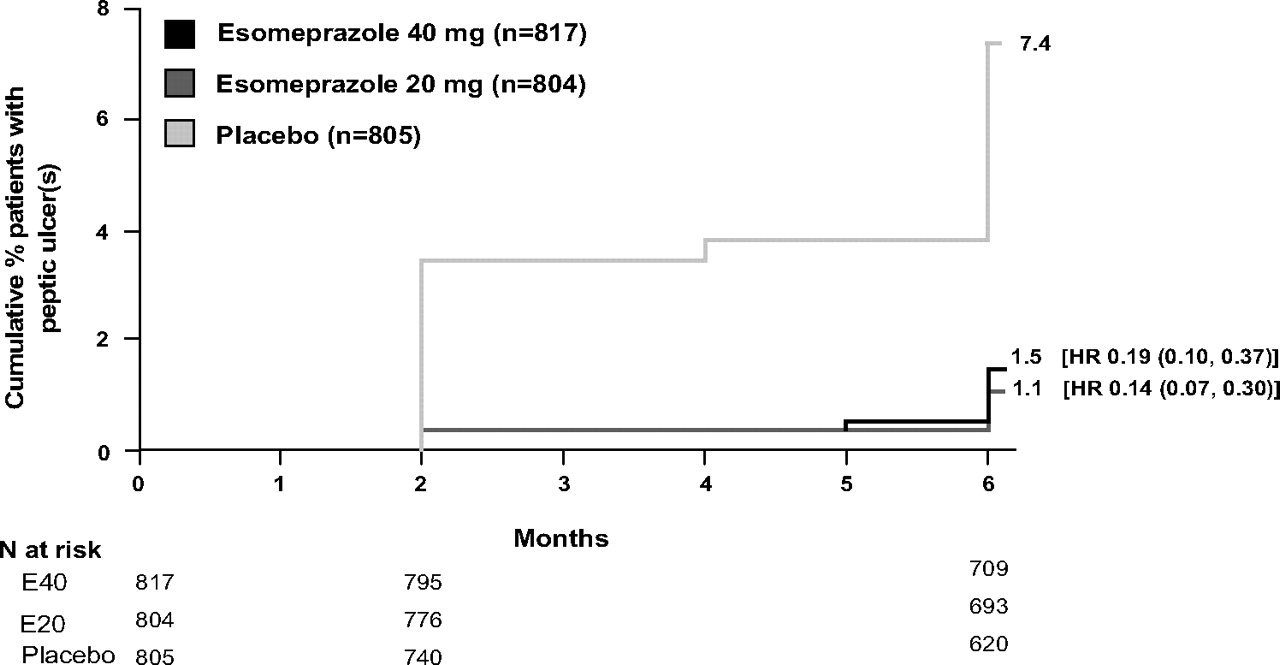
Among the ITT population, the life table estimated cumulative peptic ulcer incidence over the 26-week treatment period was 1.5% (95% CI 0.6% to 2.4%) in esomeprazole 40 mg recipients, 1.1% (95% CI 0.3% to 1.9%) in esomeprazole 20 mg recipients and 7.4% (95% CI 5.5% to 9.3%) among placebo recipients (both p<0.0001 vs placebo), resulting in an RRR versus placebo of 80% in esomeprazole 40 mg recipients and 85% in esomeprazole 20 mg recipients (figure 2), and an absolute risk reduction of 5.9% and 6.3%, respectively. The result of the corresponding per-protocol analysis, from which H pylori-positive patients were excluded, was consistent with the result of the ITT analysis. The observed incidence of peptic ulcer(s) in the ITT population was 11 esomeprazole 40 mg recipients (1.3%), 8 esomeprazole 20 mg recipients (1.0%) and 53 placebo recipients (6.6%). The peptic ulcer incidence at 26 weeks in H pylori-negative (n =1785) versus H pylori-positive (n=479) patients in the placebo arm was similar (6.8% and 5.8%, respectively). Esomeprazole 40 mg and 20 mg reduced the occurrence of peptic ulcer to <2%, relative to placebo, irrespective of H pylori status.

{kind=link}
{kind=link}
Cumulative percentage of patients with peptic ulcer(s) by week 26 (intention-to-treat population, Kaplan–Meier curve).
Overall, gastric ulcers were more prevalent than duodenal ulcers in all treatment groups. Two placebo recipients with negative and missing H pylori status, respectively, developed both duodenal and gastric ulcers. When ulcer incidence according to location was evaluated (data not shown), esomeprazole 40 mg resulted in RRRs of 74% and 90% for gastric and duodenal ulcers, respectively, versus placebo, while the respective RRRs for esomeprazole 20 mg were 83% and 90% (all p<0.0001 vs placebo; ITT population).
In total, 79% of patients used daily low-dose ASA within the range of 75–100 mg. A post hoc analysis determined that esomeprazole 40 mg and 20 mg significantly reduced peptic ulcer incidence versus placebo in patients who used ASA within this range (1.6% and 0.6% vs 6.1% of patients, respectively; p<0.0001). Similar findings were noted in patients who used ASA within the range of 101–325 mg (0.6% and 2.3% vs 8.7% of patients, respectively; p<0.02).
Safety and tolerability
Gastrointestinal complications
Few gastrointestinal complications were reported. By week 26, upper gastrointestinal complications were reported in two esomeprazole 20 mg recipients (haematemesis (n=1) and distal duodenal perforation thought to be associated with a known juxtapapillary diverticulum (n=1)), three placebo recipients (melaena (n=2) and decreased haemoglobin level (n=1)), and no esomeprazole 40 mg recipients.
Other adverse events
Overall, esomeprazole 40 mg and 20 mg were well tolerated, displaying similar tolerability profiles to placebo over 26 weeks of treatment. Adverse events were reported with a similar frequency in the three treatment groups (table 2). The most commonly reported adverse events were diarrhoea, headache and bronchitis. Overall, nine deaths occurred during the study (four esomeprazole 40 mg, four esomeprazole 20 mg and one placebo recipient); however, none of these were considered by the study investigator at the centre to be causally related to the study drug. The causes of death (as reported by study investigators) were myocardial infarction (n=2 (one esomeprazole 40 mg and one placebo recipient)), cerebrovascular accident (esomeprazole 20 mg), cardiac arrest (esomeprazole 20 mg), acute coronary syndrome (esomeprazole 20 mg), small bowel obstruction (esomeprazole 40 mg), acute renal failure (esomeprazole 40 mg), sudden death (esomeprazole 40 mg) and death from an unknown cause (esomeprazole 20 mg). The patient who died as a result of acute coronary syndrome began taking clopidogrel at the time of the cardiovascular event; none of the other patients who died were taking clopidogrel. Serious adverse events other than death were reported for 5.3% of esomeprazole 40 mg, 4.9% of esomeprazole 20 mg and 4.4% of placebo recipients; again, none of these were considered to be causally related to the study drug. There were no cardiovascular events in patients receiving clopidogrel, and no further events in patients who experienced a cardiovascular event and began taking clopidogrel during the trial (n=5). The incidence of adverse events categorised as cardiac disorders was low, and there were no clinically meaningful differences in the incidence of these events among the treatment groups (2.5%, 2.4% and 2.1% for esomeprazole 40 mg, 20 mg and placebo, respectively).
Number (%) of patients with adverse events (safety population)
Discussion
In the OBERON trial, once-daily esomeprazole 40 mg and 20 mg significantly reduced the incidence of endoscopy-confirmed peptic ulcer, compared with placebo, during 26 weeks of treatment. Esomeprazole had a similar effect among patients taking ASA between the ranges of 75–100 mg and 101–325 mg. Esomeprazole in combination with low-dose ASA was well tolerated and there were no safety concerns. The data from the OBERON trial support and extend the findings of the earlier ASTERIX trial, which demonstrated the efficacy of esomeprazole 20 mg in reducing the incidence of peptic ulcers associated with continuous use of low-dose ASA.21
The results of the OBERON trial are potentially clinically relevant in several ways. Although there is a recognised difference between endoscopic ulcers and bleeding complications, endoscopic ulcers have been used as clinical markers of gastrointestinal end points, based on the rationale that prevention of ulcer occurrence would reduce the incidence of ulcer bleeds.22 Indeed, there is evidence that concomitant PPIs decrease hospitalisations due to serious gastrointestinal bleeding in patients taking ASA or non-steroidal anti-inflammatory drugs.23 More recently, the Clopidogrel and Optimisation of Gastrointestinal Events Trial (COGENT) showed that prophylactic PPI treatment significantly reduced the rate of upper gastrointestinal bleeding among patients receiving dual antiplatelet treatment, without a significant increase in the risk of cardiovascular events.24
Peptic ulcers with the potential for gastrointestinal bleeding may influence underlying cardiovascular disease—for example, peptic ulcers can cause anaemia,25 an independent risk factor for mortality and hospitalisation in patients with myocardial infarction.26Also, gastrointestinal bleeding is independently associated with mortality and ischaemic complications in patients with acute coronary syndrome.27
The results of the OBERON trial extend the findings of ASTERIX, providing data that are relevant to clinical practice. By refining the inclusion criteria for recruiting patients with higher gastrointestinal risk than those patients who took part in ASTERIX, OBERON provides data on low-dose ASA users who are most likely to benefit from concomitant PPIs. Of note, the risk factors for gastrointestinal bleeding are also important risk factors for coronary disease.1 Patients with multiple erosions at baseline were included in the study as an increased rate of gastric/duodenal erosions was previously associated with low-dose ASA treatment.28 Patients who were naïve to low-dose ASA treatment were also included, as epidemiological data indicate that patients treated with ASA develop peptic ulcers at a higher rate during the initial treatment phase (within 1 month) than after the initial phase.11 However, our documented incidence of peptic ulcer development is still likely to be conservative; higher ulcer rates have been found in low-dose ASA users elsewhere.7 Patients with the highest gastrointestinal risk (baseline peptic ulcers, reflux oesophagitis and a history of peptic ulcer complications) and cardiovascular risk were excluded from this study for ethical reasons.
The benefit of concomitant PPI treatment in a subgroup of low-dose ASA recipients at very high gastrointestinal risk, using ulcer complications as end points, has previously been demonstrated.29 Of particular relevance to patients at increased cardiovascular risk, concomitant low-dose ASA and esomeprazole treatment significantly lowered the risk of recurrent ulcer bleeding compared with clopidogrel monotherapy.30 Given these data, it is not surprising that the use of PPIs for gastroprotection among patients with gastrointestinal risk factors who require low-dose ASA or clopidogrel for cardiovascular risk management is supported by expert opinion in the joint statement by the American College of Cardiology Foundation, American College of Gastroenterology and the American Heart Association.20 More than 20% of the ITT population were H pylori positive (as determined by a [13C]urea breath test), indicating a high level of false-negative serology screening tests at the study centres. A similar rate of false-negative tests was also noted during the ASTERIX trial. In order to preserve the gastrointestinal risk profile of the patients, the result of the confirmatory test was not provided to the investigator during the study unless requested. Since H pylori testing is not always performed in routine clinical practice before starting antiplatelet treatment, the data support the efficacy of the treatment in the target population.
Reduction of ulcer development in low-dose ASA users was recently demonstrated with use of an H2-receptor antagonist (H2-RA), famotidine, 20 mg twice daily for 3 months.31 Many methodological differences between this and our study make comparisons difficult; of particular note, over 20% of patients in the famotidine study did not have final endoscopic confirmation of ulcer status. Furthermore, 42% of placebo recipients who developed ulcers had H pylori infection, versus no instances of infection in the active treatment group, raising the possibility that many of the ulcers prevented were attributable to H pylori infection and not to low-dose ASA. Further, in a longer-term (48 weeks) trial, famotidine 40 mg/day was shown to be inferior to a PPI (pantoprazole) in the prevention of dyspeptic or bleeding ulcers/erosions in patients with ASA-related peptic ulcers/erosions.32 Concerns about the long-term use of H2-RAs for the reduction of low-dose ASA-related ulcers include tachyphylaxis, as well as lack of convincing observational data that bleeding ulcers can be reduced, leading to recommendations favouring PPI over H2-RA treatment.20 While the efficacy of H2-RA treatment in reduction of gastric bleeding has been observed, the effect was less than has been shown with PPI treatment.33 The efficacy of esomeprazole 40 mg and 20 mg was shown in an OBERON analysis for both the H pylori-negative population and patients proved to be H pylori positive after randomisation. The same observation was made in the ASTERIX study, which used esomeprazole 20 mg daily.21
In conclusion, the results of this trial demonstrate that esomeprazole 40 mg and 20 mg once daily reduces the risk of endoscopically proved peptic ulcer in patients who take low-dose ASA, and who are at risk for ulcer development. In the population studied, the esomeprazole 40 mg and 20 mg regimens appear to be equally effective.
Acknowledgments
The authors thank Anna Mett and Jackie Campbell from inScience Communications, a Wolters Kluwer business, who wrote the first draft of this paper based on direction from the authors. Medical writing services from inScience Communications were funded by AstraZeneca, the manufacturer of esomeprazole. Statistical analyses were conducted and validated by EN (AstraZeneca R&D, Mölndal, Sweden) and PJD (McMaster University, Hamilton, Ontario, Canada),observed via web-based link reproduction of the statistical analyses.
References
Footnotes
Funding This study was supported by AstraZeneca, the manufacturer of esomeprazole.
Competing interests JMS—consultant: AstraZeneca, Novartis, Pfizer, Bayer, Takeda, Pozen, NiCox. Speaker's honoraria: AstraZeneca. PJD—AstraZeneca previously supplied the study drug for a large, international, investigator-initiated, Canadian Institutes of Health Research funded randomised, controlled trial (ie, the POISE-1 Trial). JH—no conflicts of interest declared. PHK—speaker for AstraZeneca, Nycomed and Janssen-Cilag. No stocks or shares or direct financial links. AL—adviser to AstraZeneca, Pfizer, Nicox, Bayer. Speaker: AstraZeneca, Pfizer. SVvZ—grant/research support and/or honoraria: Abbott, AstraZeneca, Janssen-Ortho, Nycomed and Takeda. EN, L-ES—employees: AstraZeneca.
Ethics approval This study was conducted with the approval of the relevant local institutional review boards or independent ethics committees of each study centre, according to local regulations.
Provenance and peer review Not commissioned; externally peer reviewed.